genomation: a toolkit for annotation and visualization of genomic data
Introduction
genomation is a toolkit for annotation
and in bulk visualization of genomic features (scored or unscored)
over predefined regions.
The genomic features which the package can handle can
be anything with a minimal information of chromosome, start and end.
The features could have any length and most of the time they are
associated with a score. Typical examples of such data sets include aligned
reads from high-throughput sequencing (HTS) experiments, percent methylation
values for CpGs (or other cytosines), locations of transcription factor binding,
and so on. On the other hand, throughout the vignette we use the phrase
"genomic annotation" to refer to the regions of the genome associated with a
potential function which does not necessarily have a score
(examples: CpG islands, genes, enhancers, promoter, exons, etc. ).
These genomic annotations are usually the regions of interest, and distribution
of genomic features over/around the annotations are can make the way for
biological interpretation of the data.
The pipeline for computational knowledge extraction consists of three steps:
data filtering, integration of data from multiple sources or generation of
predictive models and biological interpretation of produced models, which leads
to novel hypotheses that can be tested in the wetlab.genomation aims
to facilitate the integration of multiple sources of genomic features with
genomic annotation or already published experimental results.
Access the data
High-throughput data which will be used to show the functonality of the
genomation are located in two places. The annotation and cap analysis
of gene expression (CAGE) data comes prepared with the genomation package, while
the raw HTS data can be found in the sister package genomationData.
To install the genomation and the complementary data package the from
the Bioconductor repository, copy and paste the following lines into your R interpreter:
biocLite('genomationData')
biocLite('genomation')
list.files(system.file('extdata',package='genomationData'))
## [1] "H1.Bisulfite-Seq.combined.chr21.bedGraph.gz"
## [2] "SamplesInfo.txt"
## [3] "wgEncodeBroadHistoneH1hescCtcfStdAlnRep1.chr21.bam"
## [4] "wgEncodeBroadHistoneH1hescCtcfStdAlnRep1.chr21.bam.bai"
## [5] "wgEncodeBroadHistoneH1hescCtcfStdPk.broadPeak.gz"
## [6] "wgEncodeBroadHistoneH1hescP300kat3bAlnRep1.chr21.bam"
## [7] "wgEncodeBroadHistoneH1hescP300kat3bAlnRep1.chr21.bam.bai"
## [8] "wgEncodeBroadHistoneH1hescP300kat3bPk.broadPeak.gz"
## [9] "wgEncodeBroadHistoneH1hescSuz12051317AlnRep1.chr21.bam"
## [10] "wgEncodeBroadHistoneH1hescSuz12051317AlnRep1.chr21.bam.bai"
## [11] "wgEncodeBroadHistoneH1hescSuz12051317Pk.broadPeak.gz"
## [12] "wgEncodeHaibTfbsA549Rad21V0422111RawRep1.chr21.bw"
## [13] "wgEncodeHaibTfbsH1hescRad21V0416102AlnRep1.chr21.bam"
## [14] "wgEncodeHaibTfbsH1hescRad21V0416102AlnRep1.chr21.bam.bai"
## [15] "wgEncodeHaibTfbsH1hescRad21V0416102PkRep1.broadPeak.gz"
## [16] "wgEncodeRikenCageA549CellPapAlnRep1.chr21.bam"
## [17] "wgEncodeRikenCageA549CellPapAlnRep1.chr21.bam.bai"
## [18] "wgEncodeSydhTfbsH1hescZnf143IggrabAlnRep1.chr21.bam"
## [19] "wgEncodeSydhTfbsH1hescZnf143IggrabAlnRep1.chr21.bam.bai"
## [20] "wgEncodeSydhTfbsH1hescZnf143IggrabPk.narrowPeak.gz"
sampleInfo = read.table(system.file("extdata/SamplesInfo.txt", package = "genomationData"),
header = TRUE, sep = "\t")
sampleInfo[1:5, 1:5]
## fileName dataOrigin
## 1 wgEncodeBroadHistoneH1hescCtcfStdAlnRep1.chr21.bam Encode
## 2 wgEncodeBroadHistoneH1hescP300kat3bAlnRep1.chr21.bam Encode
## 3 wgEncodeBroadHistoneH1hescSuz12051317AlnRep1.chr21.bam Encode
## 4 wgEncodeHaibTfbsH1hescRad21V0416102AlnRep1.chr21.bam Encode
## 5 wgEncodeSydhTfbsH1hescZnf143IggrabAlnRep1.chr21.bam Encode
## cellType sampleName experimentType
## 1 H1hesc Ctcf ChipSeq
## 2 H1hesc P300 ChipSeq
## 3 H1hesc Suz12 ChipSeq
## 4 H1hesc Rad21 ChipSeq
## 5 H1hesc Znf143 ChipSeq
library(genomation)
data(cage)
data(cpgi)
list.files(system.file("extdata", package = "genomation"))
Data input
One of larger hindrances in computational genomics stems from the myriad of
formats that are used to store the data. Although some formats have been
selected as de facto standards for specific kind of biological data (e.g. BAM, VCF),
almost all publications come with supplementary tables that do not have the
same structure, but hold similar information. The tables usually have a tabular
format, contain the locationof elements in genomic coordinates and various
metadata colums. genomation contais functions to read genomic
features and genomic annotation provided they are in a tabular format.
These functions will read the data from flat files into GRanges or GRangesList
objects.
readGeneric is the workhorse of the genomation package. It is a function
developed specifically for input of genomic data in tabular formats, and their
conversion to a GRanges object.
By default, the function persumes that the file is a standard .bed file
containing columns chr, start, end.
library(genomation)
tab.file1 = system.file("extdata/tab1.bed", package = "genomation")
readGeneric(tab.file1)
## GRanges object with 6 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr21 [9437272, 9439473] *
## [2] chr21 [9483485, 9484663] *
## [3] chr21 [9647866, 9648116] *
## [4] chr21 [9708935, 9709231] *
## [5] chr21 [9825442, 9826296] *
## [6] chr21 [9909011, 9909218] *
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
readGeneric(tab.file1, keep.all.metadata = TRUE)
## GRanges object with 6 ranges and 3 metadata columns:
## seqnames ranges strand | V4 V5 V6
## <Rle> <IRanges> <Rle> | <integer> <integer> <numeric>
## [1] chr21 [9437272, 9439473] * | 285 1426 25.9
## [2] chr21 [9483485, 9484663] * | 165 818 28
## [3] chr21 [9647866, 9648116] * | 18 168 14.4
## [4] chr21 [9708935, 9709231] * | 31 218 20.9
## [5] chr21 [9825442, 9826296] * | 120 568 28.1
## [6] chr21 [9909011, 9909218] * | 20 143 19.3
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
readGeneric(tab.file1, meta.col = list(CpGnum = 4))
## GRanges object with 6 ranges and 1 metadata column:
## seqnames ranges strand | CpGnum
## <Rle> <IRanges> <Rle> | <integer>
## [1] chr21 [9437272, 9439473] * | 285
## [2] chr21 [9483485, 9484663] * | 165
## [3] chr21 [9647866, 9648116] * | 18
## [4] chr21 [9708935, 9709231] * | 31
## [5] chr21 [9825442, 9826296] * | 120
## [6] chr21 [9909011, 9909218] * | 20
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
readGeneric(tab.file1, header = TRUE, keep.all.metadata = TRUE)
## GRanges object with 5 ranges and 3 metadata columns:
## seqnames ranges strand | X285 X1426 X25.9
## <Rle> <IRanges> <Rle> | <integer> <integer> <numeric>
## [1] chr21 [9483485, 9484663] * | 165 818 28
## [2] chr21 [9647866, 9648116] * | 18 168 14.4
## [3] chr21 [9708935, 9709231] * | 31 218 20.9
## [4] chr21 [9825442, 9826296] * | 120 568 28.1
## [5] chr21 [9909011, 9909218] * | 20 143 19.3
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
tab.file2 = system.file("extdata/tab2.bed", package = "genomation")
readGeneric(tab.file2, chr = 3, start = 2, end = 1)
## GRanges object with 6 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr21 [9437272, 9439473] *
## [2] chr21 [9483485, 9484663] *
## [3] chr21 [9647866, 9648116] *
## [4] chr21 [9708935, 9709231] *
## [5] chr21 [9825442, 9826296] *
## [6] chr21 [9909011, 9909218] *
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
gff.file = system.file("extdata/chr21.refseq.hg19.gtf", package = "genomation")
gff = gffToGRanges(gff.file)
head(gff)
## GRanges object with 6 ranges and 5 metadata columns:
## seqnames ranges strand | source feature
## <Rle> <IRanges> <Rle> | <character> <character>
## [1] chr21 [9825832, 9826011] + | hg19_refGene exon
## [2] chr21 [9826203, 9826263] + | hg19_refGene exon
## [3] chr21 [9907189, 9907492] - | hg19_refGene exon
## [4] chr21 [9909047, 9909277] - | hg19_refGene exon
## [5] chr21 [9966322, 9966380] - | hg19_refGene exon
## [6] chr21 [9968516, 9968593] - | hg19_refGene exon
## score frame group
## <numeric> <character> <character>
## [1] 0 . gene_id NR_037421; transcript_id NR_037421;
## [2] 0 . gene_id NR_037458; transcript_id NR_037458;
## [3] 0 . gene_id NR_038328; transcript_id NR_038328;
## [4] 0 . gene_id NR_038328; transcript_id NR_038328;
## [5] 0 . gene_id NR_038328; transcript_id NR_038328;
## [6] 0 . gene_id NR_038328; transcript_id NR_038328;
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
gff = gffToGRanges(gff.file, split.group = TRUE)
head(gff)
# reading genes stored as a BED files
cpgi.file = system.file("extdata/chr21.CpGi.hg19.bed", package = "genomation")
cpgi.flanks = readFeatureFlank(cpgi.file)
head(cpgi.flanks$flanks)
## GRanges object with 6 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr21 [9823442, 9825441] *
## [2] chr21 [9826297, 9828296] *
## [3] chr21 [9907011, 9909010] *
## [4] chr21 [9909219, 9911218] *
## [5] chr21 [9966264, 9968263] *
## [6] chr21 [9968621, 9970620] *
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
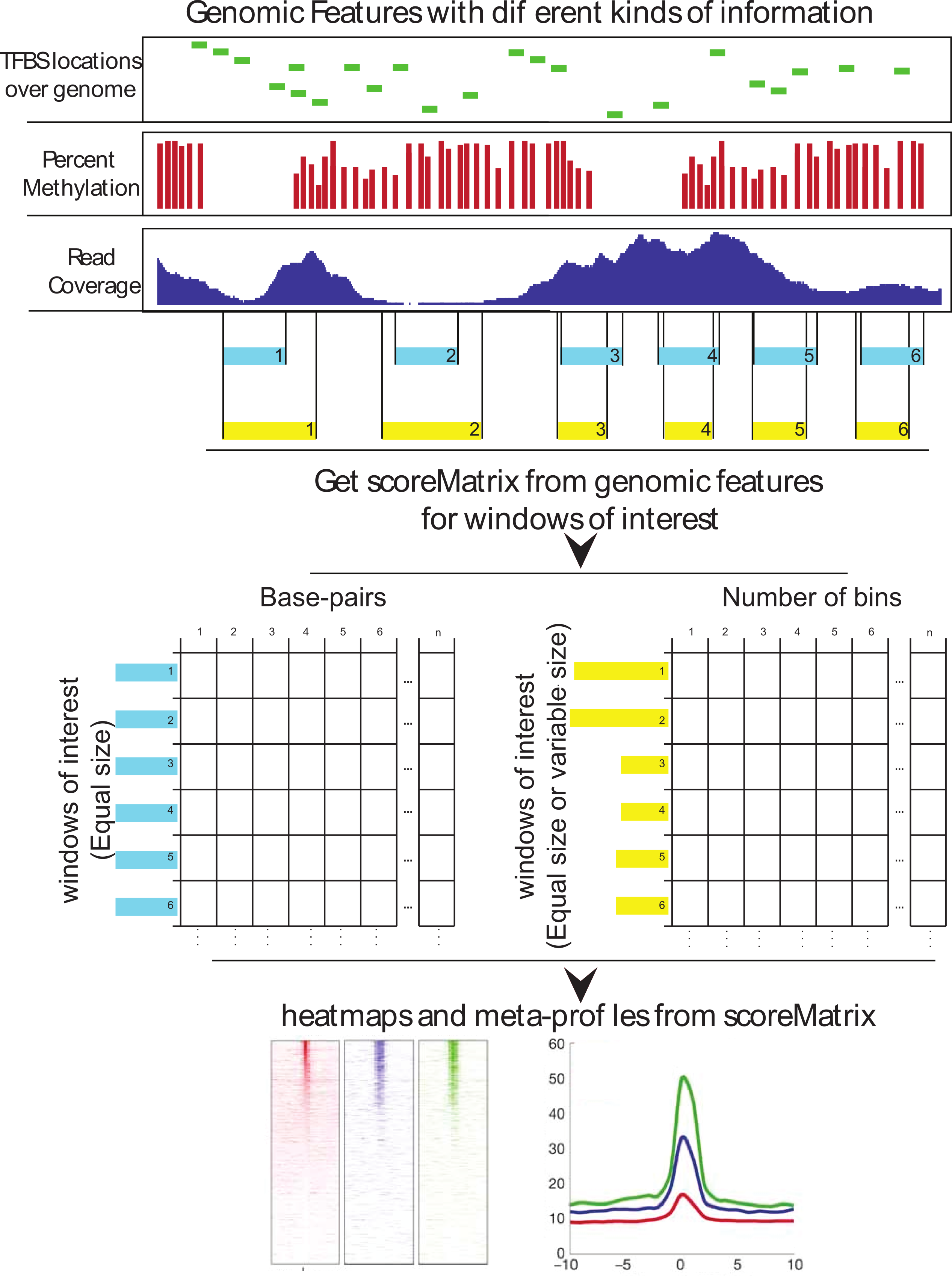
Extraction and visualization of genomic data
A standard step in a computational genomics experiment is visualization of
average enrichment over a certain predefined set of ranges, such as mean coverage
of a certain histone modification around a transcription factor binding site,
or visualization of histone positions around transcription start sites.
Extraction of data over genomic winows
ScoreMatrix and ScoreMatrixBin are functions used to extract
data over predefined windows.
ScoreMatrix is used when all of the windows
have the same width, such as a designated area around the transcription start
site, while the ScoreMatrixBin is designed for use with windows of
unequal width (e.g. enrichment of methylation over exons).
Both functions have 2 main arguments: target and
windows. target is the data that we want to extract, while the
windows represents the regions over which we want to see the enrichment.
The target data can be in 3 forms: a GRanges, a RLeList or a path to an indexed
.bam file. The windows must be GRanges object.
As an example we will extract the density of cage tags around the promoters on
the human chromosome 21.
data(cage)
data(promoters)
sm = ScoreMatrix(target = cage, windows = promoters)
sm
## scoreMatrix with dims: 1055 2001
data(cage)
gff.file = system.file("extdata/chr21.refseq.hg19.gtf", package = "genomation")
exons = gffToGRanges(gff.file, filter = "exon")
sm = ScoreMatrixBin(target = cage, windows = exons, bin.num = 50)
sm
data(promoters)
data(cpgi)
data(cage)
cage$tpm = NULL
targets = list(cage = cage, cpgi = cpgi)
sm = ScoreMatrixList(targets = targets, windows = promoters, bin.num = 50)
## working on: cage
## working on: cpgi
## scoreMatrixlist of length:2
##
## 1. scoreMatrix with dims: 1055 50
## 2. scoreMatrix with dims: 1055 50
Visualization of multiple genomic experiments
There are 2 basic modes of visualization of enrichment over windows: either
as a heatmap, or as a histogram. heatMatrix, plotMeta and
multiHeatMatrix are functions for visualization of ScoreMatrix
and ScoreMatrixList objects.
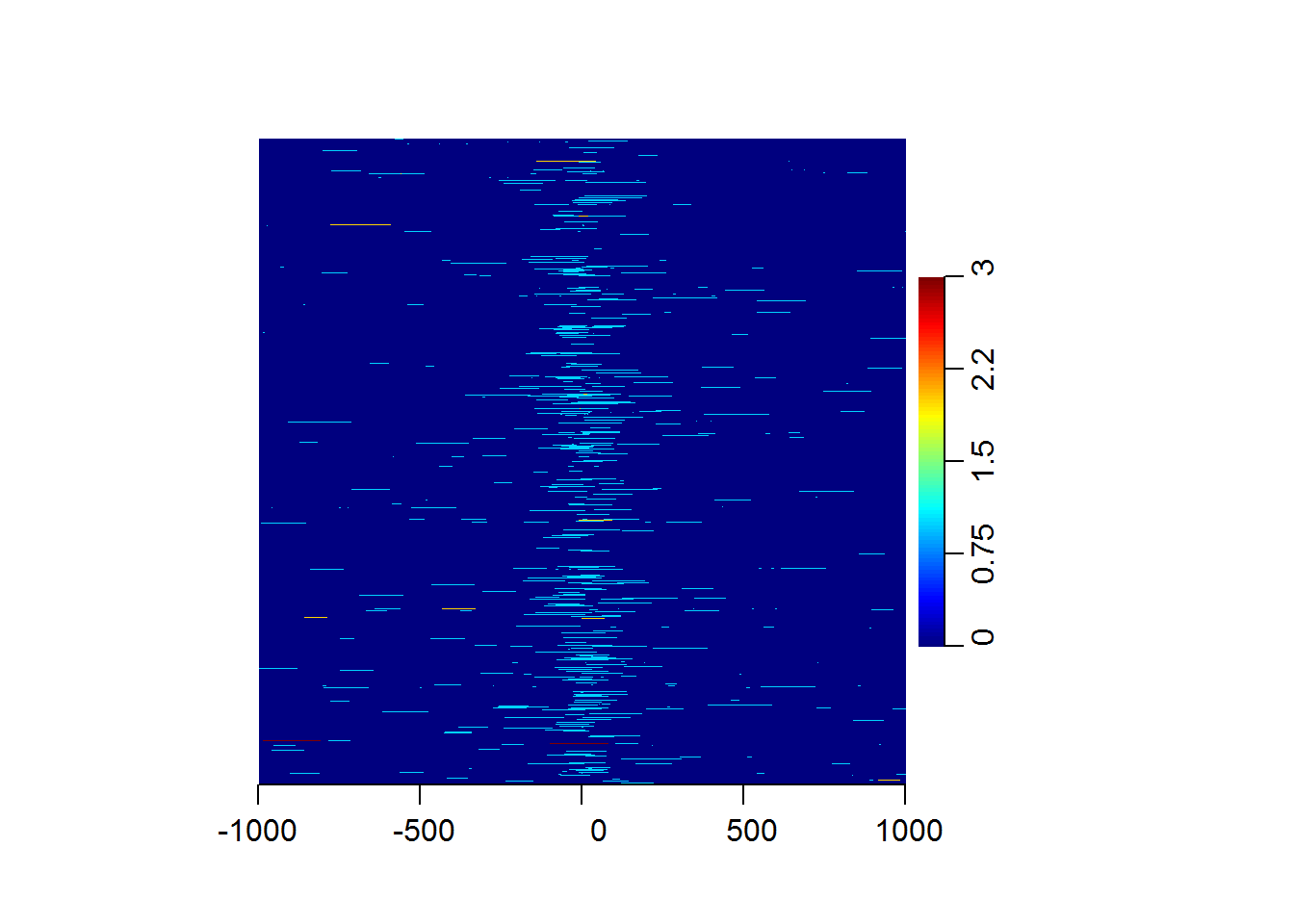
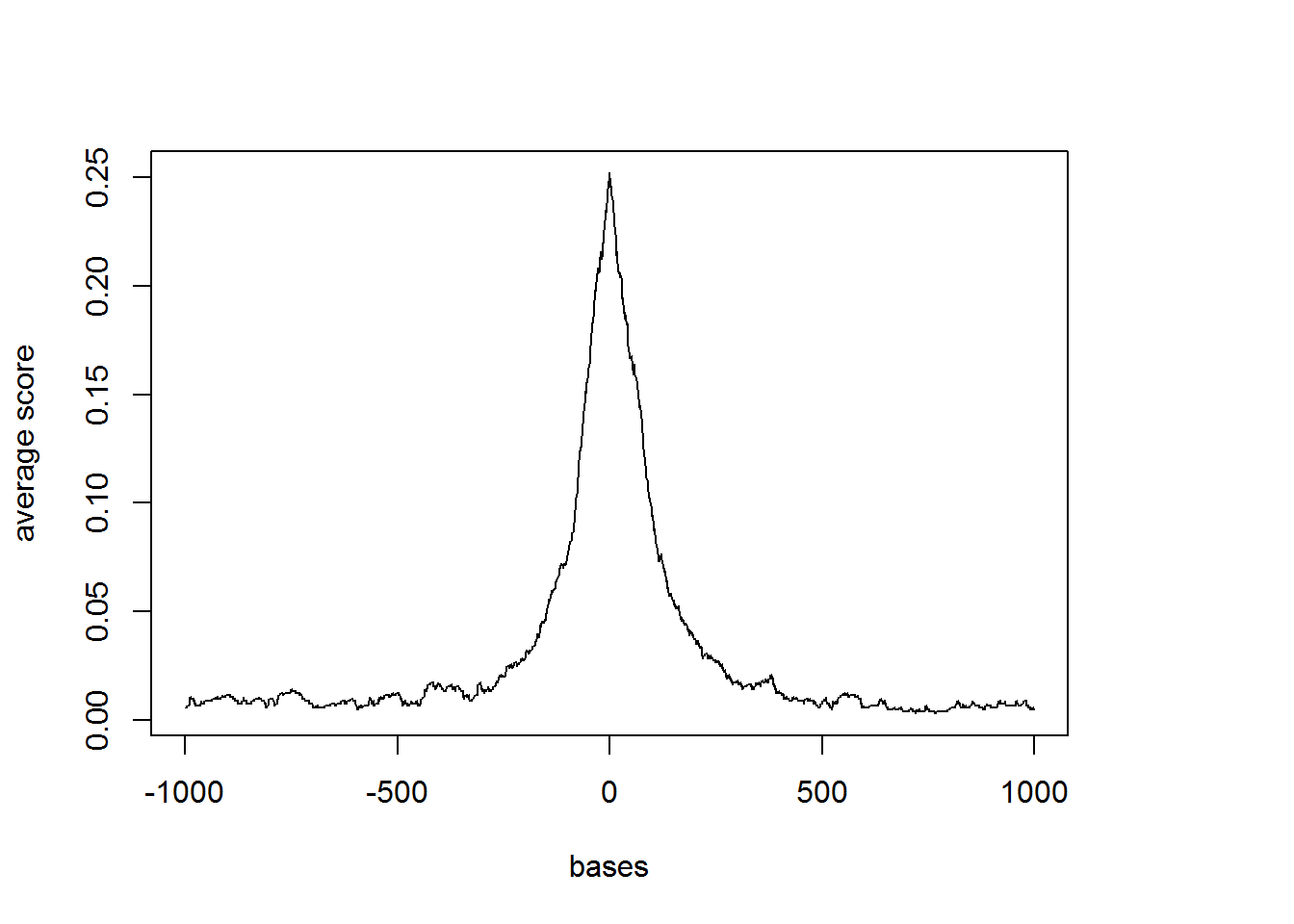
We will plot the distribution of CAGE tags around promoters on human chr21.
data(cage)
data(promoters)
sm = ScoreMatrix(target = cage, windows = promoters)
heatMatrix(sm, xcoords = c(-1000, 1000))
plotMeta(sm, xcoords = c(-1000, 1000))
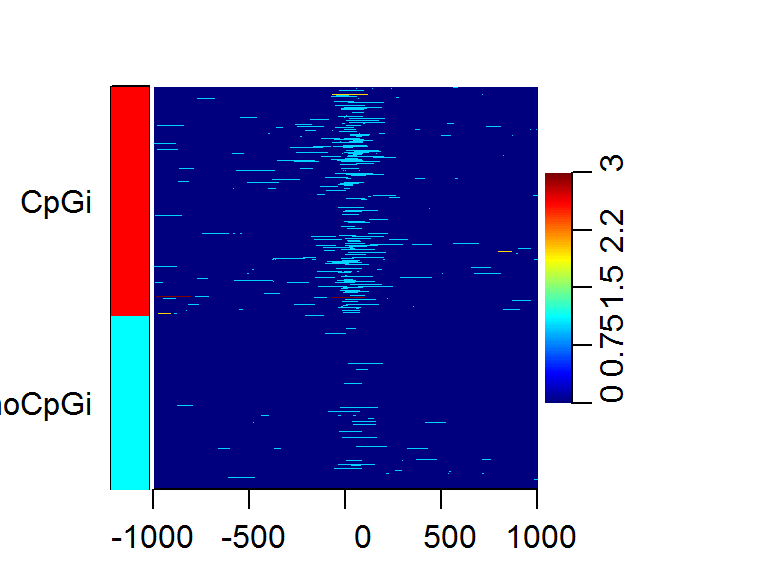
The heatMatrix function can also take a list of numeric vectors
designating row names, or a factor variable that represent our
annotation over the windows.
data(cage)
data(promoters)
data(cpgi)
sm = ScoreMatrix(target = cage, windows = promoters, strand.aware = TRUE)
cpg.ind = which(countOverlaps(promoters, cpgi) > 0)
nocpg.ind = which(countOverlaps(promoters, cpgi) == 0)
heatMatrix(sm, xcoords = c(-1000, 1000), group = list(CpGi = cpg.ind, noCpGi = nocpg.ind))
Because the enrichment in windows can have a high dynamic range, it is sometimes
convenient to scale the matrix before plotting.
sm.scaled = scaleScoreMatrix(sm)
heatMatrix(sm.scaled, xcoords = c(-1000, 1000), group = list(CpGi = cpg.ind,
noCpGi = nocpg.ind))
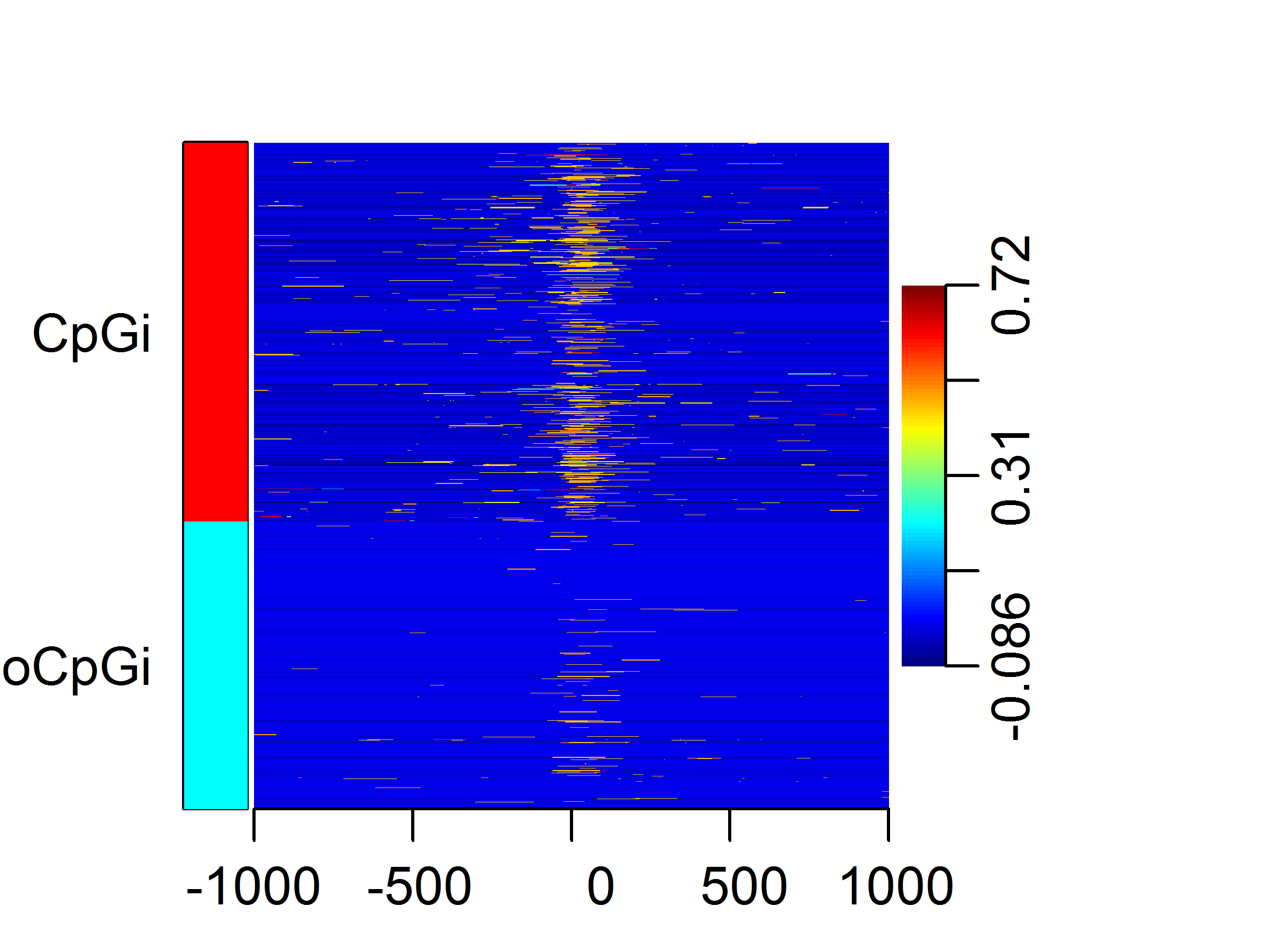
Several experiments can be plotted in a side by side fashion using a combination
of ScoreMatrixList and multiHeatMatrix.
cage$tpm = NULL
targets = list(cage = cage, cpgi = cpgi)
sml = ScoreMatrixList(targets = targets, windows = promoters, bin.num = 50,
strand.aware = TRUE)
multiHeatMatrix(sml, xcoords = c(-1000, 1000))
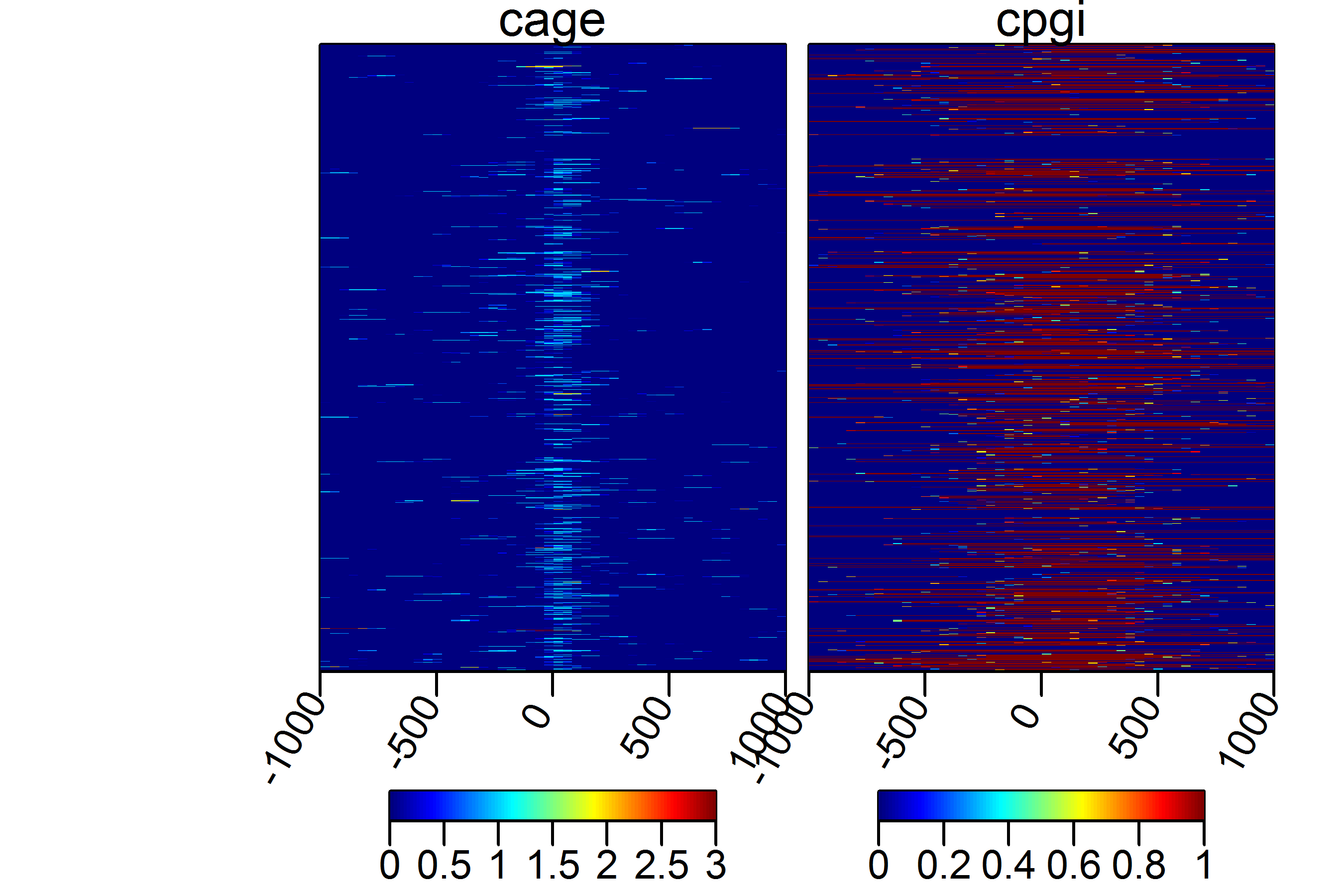
We can put the kmeans=TRUE to see whether there are any patterns present in the data.
multiHeatMatrix(sml, xcoords = c(-1000, 1000), kmeans = TRUE, k = 2)
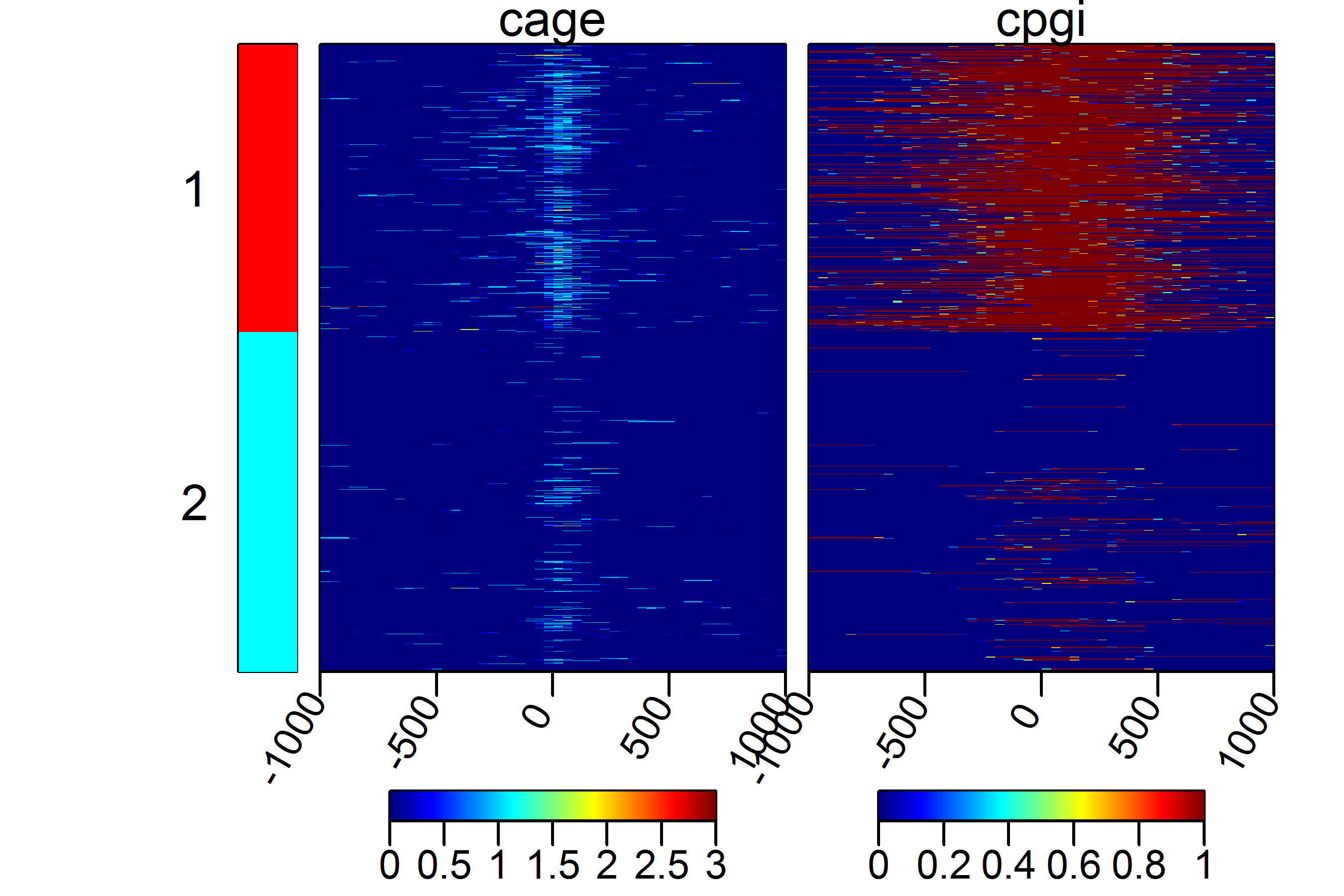
More advance usage of the ScoreMatrix family of functions and their
visualtization can be found in the specific use-cases at the end of the vignette.
Annotation of genomic features
Searching for correlation between sets of genomic features is a standard exploratory
method in computational genomics. It is usually done by looking at the overlap between
2 or more sets of ranges and calculating various overlap statistics.
genomation contains two sets of functions for annotation of ranges:
the first one is used to facilitate the general annotation of any sets of ranges,
while the second one is used to annotate a given feature with gene structures (promoter,
exon, intron).
Annotation by generic features
Firstly, we will select the broadPeak files from the genomatonData package,
and read in the peaks for the Ctcf transcription factor
library(genomationData)
genomationDataPath = system.file("extdata", package = "genomationData")
sampleInfo = read.table(file.path(genomationDataPath, "SamplesInfo.txt"), header = TRUE,
sep = "\t", stringsAsFactors = FALSE)
peak.files = list.files(genomationDataPath, full.names = TRUE, pattern = "broadPeak")
names(peak.files) = sampleInfo$sampleName[match(basename(peak.files), sampleInfo$fileName)]
ctcf.peaks = readBroadPeak(peak.files["Ctcf"])
Now we will annotate the human the Ctcf binding sites using the CpG islands.
Because the CpG islands are restricted to chromosomes 21 and 22, we will set the
intersect.chr = TRUE, which will limit the analysis only to the
chromosomes that are present in both data sets.
data(cpgi)
peak.annot = annotateWithFeature(ctcf.peaks, cpgi, intersect.chr = TRUE)
## intersecting chromosomes...
## summary of target set annotation with feature annotation:
## Rows in target set: 3964
## ----------------------------
## percentage of target elements overlapping with features:
## cpgi other
## 7.62 92.38
##
## percentage of feature elements overlapping with target:
The output of the annotateWithFeature function shows three types of information:
The total number of elements in the target dataset, the percentage of target
dataset that overlaps with the feature dataset. And the percentage of the feature
elements that overlap the target.
Annotation of genomic features by gene structures
To find the distribution of our designated features around gene structures,
we will first read the transcript features from a file using the
readTranscriptFeatures function. readTranscriptFeatures reads
a bed12 formatted file and parses the coordinates into a GRangesList containing
four elements: exons, introns. promoters and transcription start sites (TSSes).
bed.file = system.file("extdata/chr21.refseq.hg19.bed", package = "genomation")
gene.parts = readTranscriptFeatures(bed.file)
## Reading the table...
## Calculating intron coordinates...
## Calculating exon coordinates...
## Calculating TSS coordinates...
## Calculating promoter coordinates...
## Outputting the final GRangesList...
annotateWithGeneParts will give us the overlap statistics between our
CTCF peaks and gene structures. We will again use the intersect.chr=TRUE
to limit the analysis.
ctcf.annot = annotateWithGeneParts(ctcf.peaks, gene.parts, intersect.chr = TRUE)
## intersecting chromosomes...
## Summary of target set annotation with genic parts
## Rows in target set: 1681
## -----------------------
## percentage of target features overlapping with annotation:
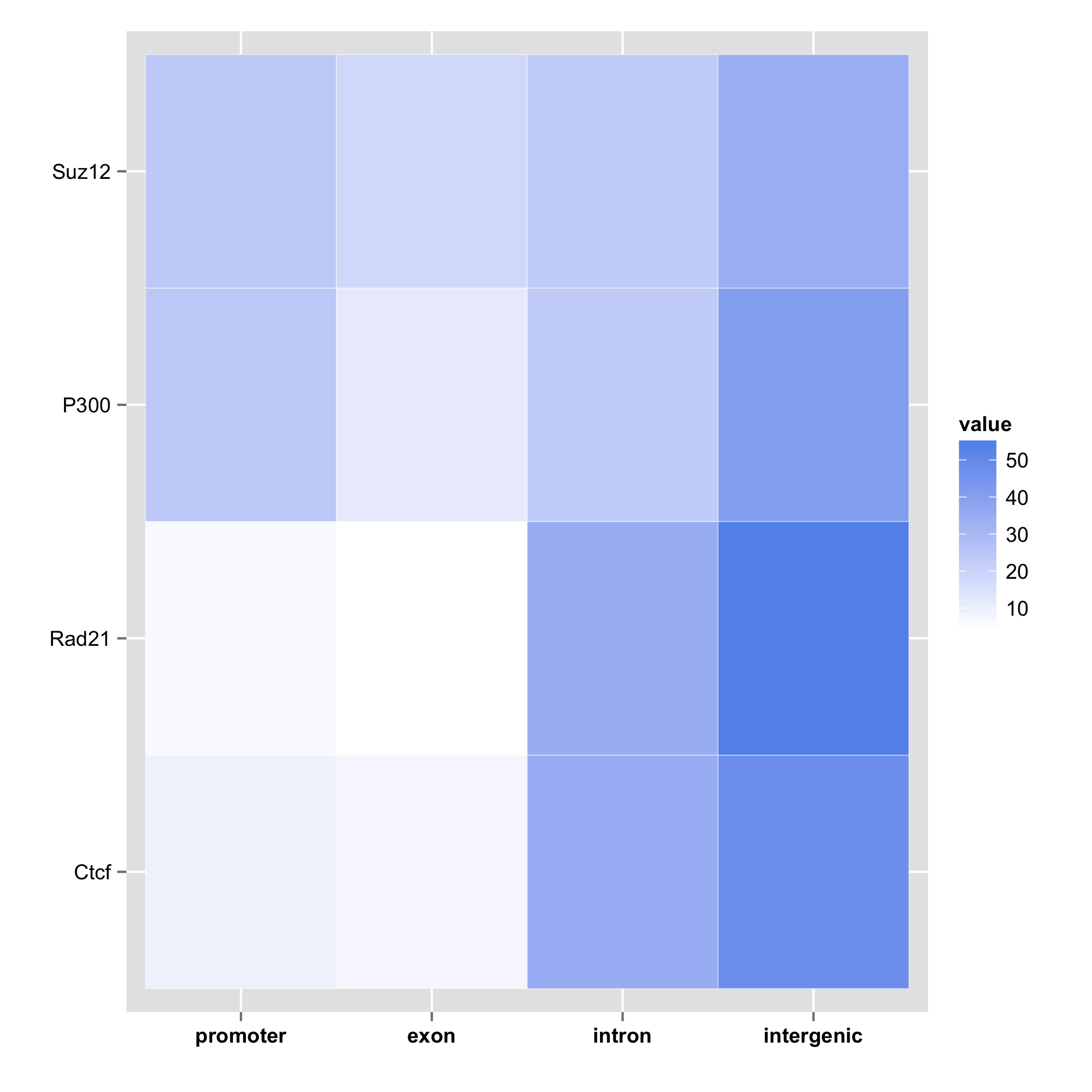
## promoter exon intron intergenic
## 9.58 13.50 47.65 47.17
##
## percentage of target features overlapping with annotation:
## (with promoter > exon > intron precedence):
## promoter exon intron intergenic
## 9.58 7.91 35.34 47.17
##
## percentage of annotation boundaries with feature overlap:
## promoter exon intron
## 38.36 15.40 29.33
##
## summary of distances to the nearest TSS:
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0 7977 28470 66060 74690 1195000
annotateWithGeneParts can also take a set of feature ranges as an argument.
We will use the readGeneric function to load all of the broadPeak files in the
genomationData, which we will then annotate.
peaks = GRangesList(lapply(peak.files, readGeneric))
names(peaks) = names(peak.files)
annot.list = annotateWithGeneParts(peaks, gene.parts, intersect.chr = TRUE)
## Working on: Ctcf
## intersecting chromosomes...
## Working on: P300
## intersecting chromosomes...
## Working on: Suz12
## intersecting chromosomes...
## Working on: Rad21
## intersecting chromosomes...
Gene annotation of multiple feature objects can be visualized in a form of a heatmap,
where rows represent samples, columns the gene structure, and the value is the
percentage of overlap given by priority. If cluster=TRUE, then the function
will use hierarhical clustering to order the heatmap.
plotGeneAnnotation(annot.list)
plotGeneAnnotation(annot.list, cluster = TRUE)
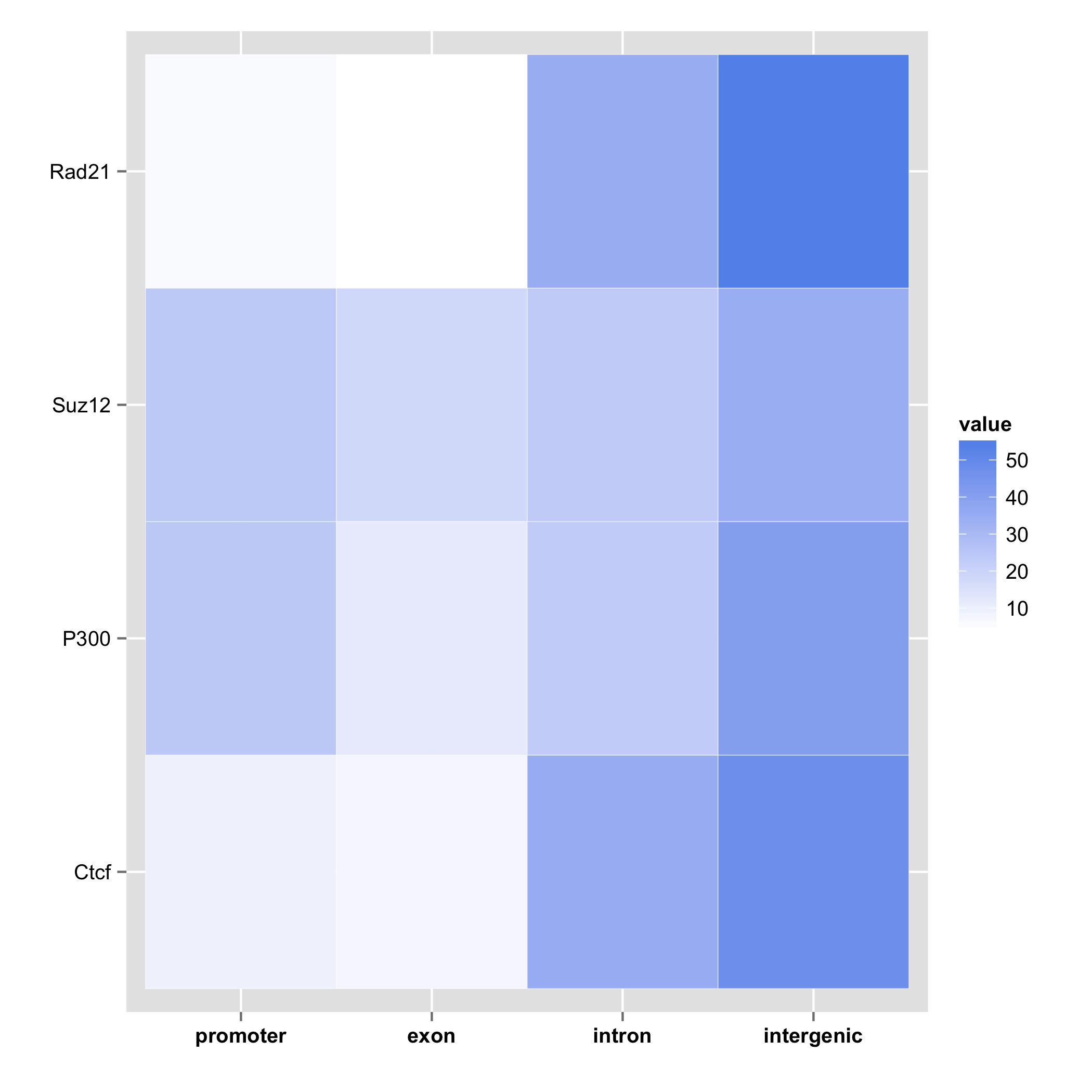
Use cases for genomation package
The genomation package provides generalizable functions for genomic data analysis
and visualization. Below we will demonstrate the functionality on specific use cases
Visualization of ChiP sequencing data
We will visualize the binding profiles of 6 transcription factors around the
Ctcf binding sites.
In the fist step we will select the *.bam files containing mapped reads.
genomationDataPath = system.file("extdata", package = "genomationData")
bam.files = list.files(genomationDataPath, full.names = TRUE, pattern = "bam$")
bam.files = bam.files[!grepl("Cage", bam.files)]
Firstly, we will read in the Ctcf peaks, filter regions from human chromosome 21,
and order them by their signal values. In the end we will resize all ranges to
have a uniform width of 500 bases, fixed on the center of the peak.
ctcf.peaks = readBroadPeak(file.path(genomationDataPath, "wgEncodeBroadHistoneH1hescCtcfStdPk.broadPeak.gz"))
ctcf.peaks = ctcf.peaks[seqnames(ctcf.peaks) == "chr21"]
ctcf.peaks = ctcf.peaks[order(-ctcf.peaks$signalValue)]
ctcf.peaks = resize(ctcf.peaks, width = 1000, fix = "center")
In order to extract the coverage values of all transcription factors around
chipseq peaks, we will use the ScoreMatrixList function.
ScoreMatrixList assign names to each element of the list based on the
names of the bam files. We will use the names of the files to find the
corresponding names of each sample in the SamplesInfo.txt.
Using the heatmapProfile on our ScoreMatrixList,
we can plot the underlying signal side by side.
sml = ScoreMatrixList(bam.files, ctcf.peaks, bin.num = 50, type = "bam")
sampleInfo = read.table(system.file("extdata/SamplesInfo.txt", package = "genomationData"),
header = TRUE, sep = "\t")
names(sml) = sampleInfo$sampleName[match(names(sml), sampleInfo$fileName)]
multiHeatMatrix(sml, xcoords = c(-500, 500), col = c("lightgray", "blue"))
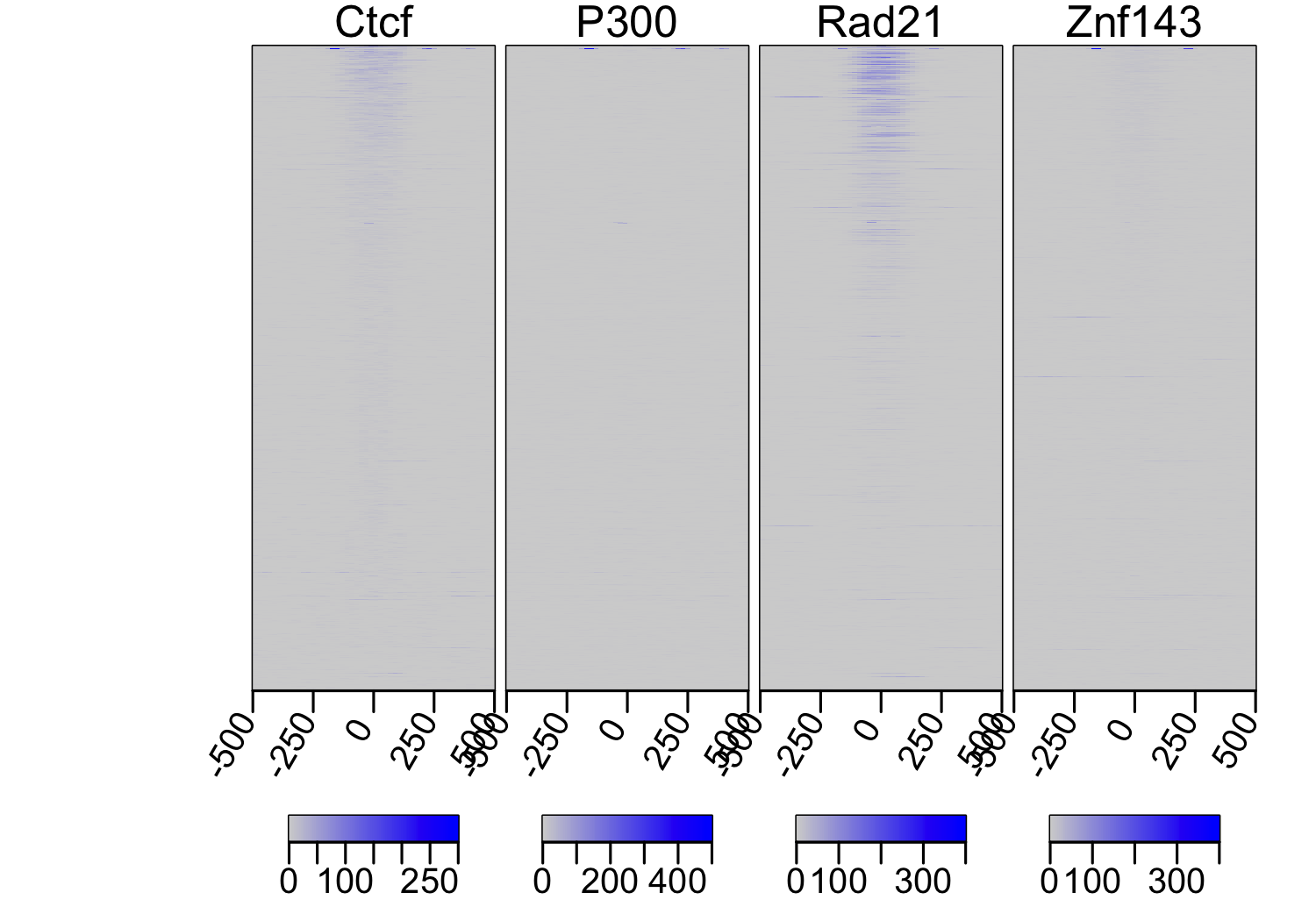 Heatmap profile of unscaled coverage shows a slight colocalization of
Ctcf, Rad21 and Znf143.
Heatmap profile of unscaled coverage shows a slight colocalization of
Ctcf, Rad21 and Znf143.
Because of the large range of signal values in chipseq peaks, the heatmapProfile
will not show the true extent of colocalization. To get around this, it is advisable
to independently scale the rows of each element in the ScoreMatrixList.
sml.scaled = scaleScoreMatrixList(sml)
multiHeatMatrix(sml.scaled, xcoords = c(-500, 500), col = c("lightgray", "blue"))
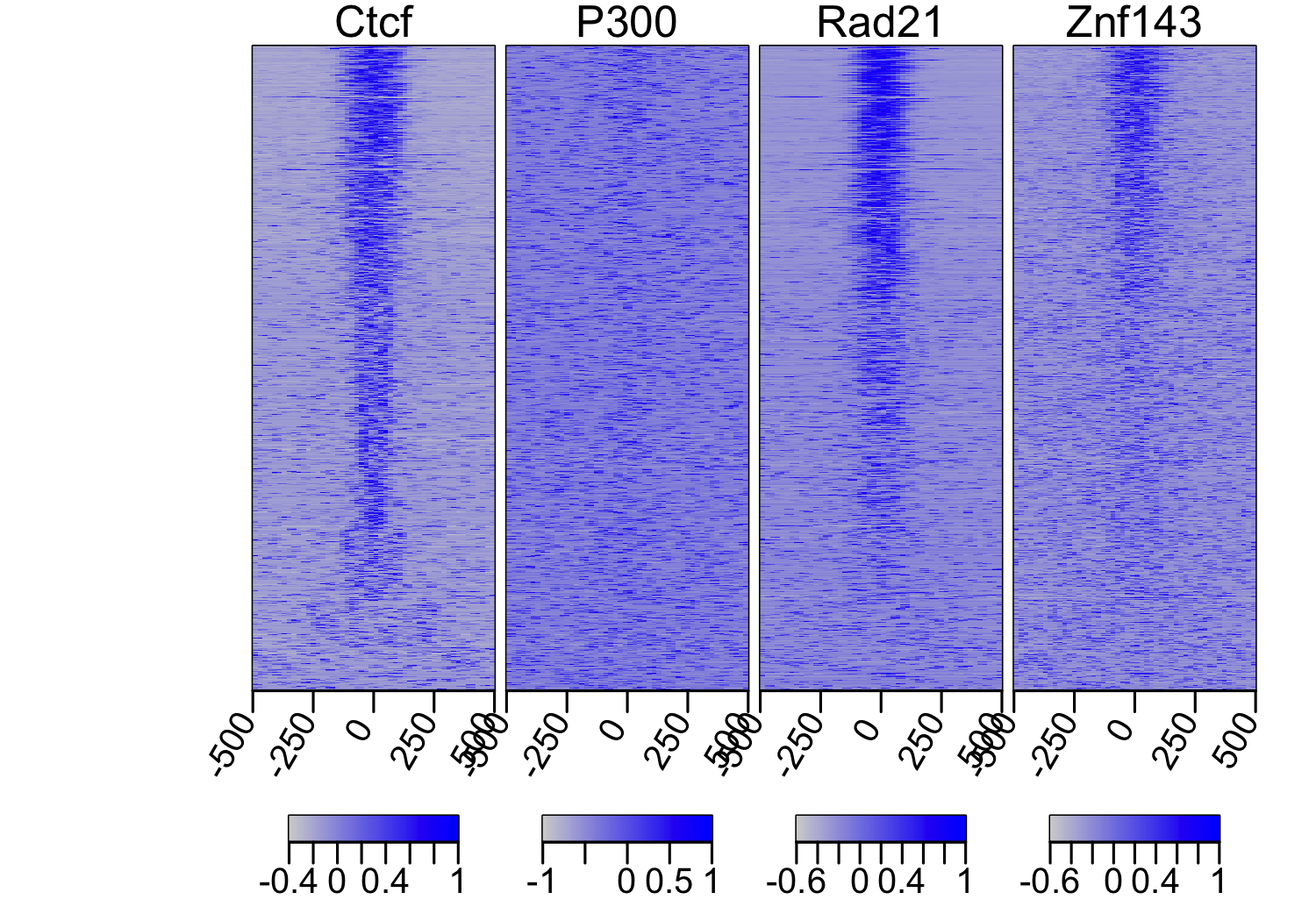 Heatmap profile of scaled coverage shows much stronger colocalization
of the transcription factors; nevertheless, it is evident that some of the CTCF
peaks have a very weak enrichment.
Heatmap profile of scaled coverage shows much stronger colocalization
of the transcription factors; nevertheless, it is evident that some of the CTCF
peaks have a very weak enrichment.
Combinatorial binding of transcription factors
In the first step we will read all peak files into a GRanges list. We will use
the SamplesInfo file from the genomationData to get he names of the
samples. Four of the peak files are in the Encode broadPeak format, while one is
in the narrowPeak. To read the files, we will use the readGeneric
function. It enables us to select from the files only columns of interest.
As the last step, we will restrict ourselves to peaks that are located on
chromosome 21 and have width 100 and 1000 bp
genomationDataPath = system.file("extdata", package = "genomationData")
sampleInfo = read.table(file.path(genomationDataPath, "SamplesInfo.txt"), header = TRUE,
sep = "\t", stringsAsFactors = FALSE)
peak.files = list.files(genomationDataPath, full.names = TRUE, pattern = "Peak.gz$")
peaks = list()
for (i in 1:length(peak.files)) {
file = peak.files[i]
name = sampleInfo$sampleName[match(basename(file), sampleInfo$fileName)]
message(name)
peaks[[name]] = readGeneric(file, meta.col = list(score = 5))
}
## Ctcf
## P300
## Suz12
## Rad21
## Znf143
peaks = GRangesList(peaks)
peaks = peaks[seqnames(peaks) == "chr21" & width(peaks) < 1000 & width(peaks) >
100]
To find the combination of binding sites we will use the findFeatureComb
function. It takes a granges list object, finds the union of the
ranges and designates each range by the combination of overlaps from the original set.
By default, the returned ranges will have a numeric class
meta data column, which designates the correponding combination.
If you are interested in the names of the TF which make
the combinations, put the use.names=TRUE.
tf.comb = findFeatureComb(peaks, width = 1000)
To visualize the results, we will plot the enricment of resulting regions.
Before doing that we will order the regions by their class argument.
tf.comb = tf.comb[order(tf.comb$class)]
bam.files = list.files(genomationDataPath, full.names = TRUE, pattern = "bam$")
bam.files = bam.files[!grepl("Cage", bam.files)]
sml = ScoreMatrixList(bam.files, tf.comb, bin.num = 20, type = "bam")
names(sml) = sampleInfo$sampleName[match(names(sml), sampleInfo$fileName)]
sml.scaled = scaleScoreMatrixList(sml)
multiHeatMatrix(sml.scaled, xcoords = c(-500, 500), col = c("lightgray", "blue"))
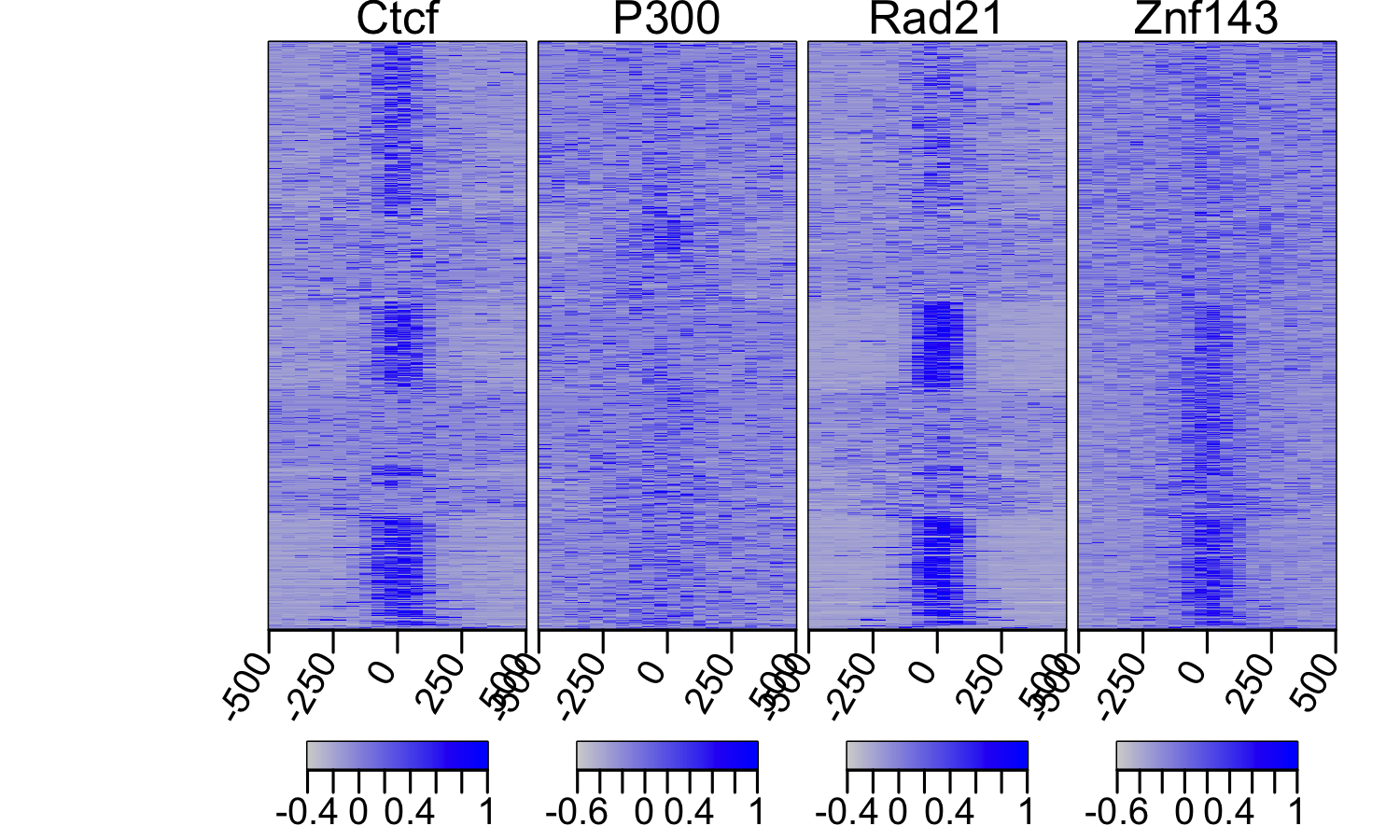
The plot shows perfectly how misleading the peak calling process can be.
Although the plots show that CTFC, Rad21 and Znf143 have almost perfect
colozalization, peak callers have trouble identifying peaks in regions with
lower enrichments and as a result, we get different statistics
when using overlaps.
Using data from AnnotationHub
We can also use data from AnnotationHub since it can return data as GRanges object. Below we download CpG island and DNAse peak locations from AnnotationHub and get a scoreMatrix on the CpG islands.
library(AnnotationHub)
ah = AnnotationHub()
# retrieve CpG island data from annotationHub
cpgi = ah$goldenpath.hg19.database.cpgIslandExt_0.0.1.RData
dnase = paste("goldenpath", "hg19.encodeDCC", "wgEncodeOpenChromDnase", "wgEncodeOpenChromDnase8988tPk",
"narrowPeak_0.0.1.RData", sep = ".")
dnaseP = ah[[dnase]]
sm = ScoreMatrixBin(target = dnaseP, windows = cpgi, strand.aware = FALSE)