Comparative Identification and Assessment of Single Nucleotide Variants Through Shifts in Base Call Frequencies
Table of Contents
1 Motivation
The advances in high-throughput nucleotide sequencing have lead to new possibilities of identifying genomic alterations, which is of particular interest for cancer research. While the detection of genomic variants is often conducted by comparing against a population-based reference sequence (such as the human GRCh 37), in the investigation of tumors a comparison between matched samples is predominantly pursued. Considering a test sample \(T\) and a control sample \(C\), the aim is to identify single nucloetide variants (SNVs) which show significant changes between the two. Typically, this is applied for comparing a tumor sample against a matched normal/constitutive sample of the same patient, focusing on alterations that may have occurred in the transition to the cancerous tissue, and therefore often termed somatic variants. However, a broader range of questions can be addressed by extending the focus to include genomic changes of temporally or spatially separated samples to each other, to investigate tumor evolution and subclonal shifts [11].
Extending the scope of comparative variant analyses in a cancer related setting may require an accompanying change in definitions. In the classical tumor versus normal setting, a somatic event is characterized by both the presence of the variant in the tumor and the absence in the matched normal [17]. This definition is not satisfactory for comparing different tumor stages against each other. If we imagine two stages of a tumor haboring multiple subclones, variants may be present in both stages with changing abundances. Here, the classical definition of a somatic variant would result in missing the event. In the following, we will therefore focus on estimating the shift in variant allele frequencies between a test and a control samples. The classical somatic definition requiring the variant to be absent in the control constitutes a special case of this.
Numerous approaches focus on more narrowly defined somatic comparisons, motivated by scientific questions in the field of cancer genomics [16] [17]. Most existing methods do not allow an explicit statement about the evidence for the absence of a putative variant in a sample, and a clear distinction is needed whether a negative call results from a true negative site or the lack of statistical power to detect it. As an example, image a variant calling approach using a hypothesis test with the null hypothesis that the variant frequencies at a locus are identical for both samples. While a significant p-value provides evidence for the presence of a variant, inverting this argument does not work: A non-significant p-value can both be indicative of the absence or insufficient power. Furthermore, most methods return only a point estimate for the variant frequency in the tumor, without stating the confidence of the estimate. However, this information would be critical to reliably differentiate between variants arising from competing subclonal populations.
With the Rariant package, we pursue a generalized approach for identifying and
quantifying SNV alterations from high-throughput sequencing data in comparative
settings. By focusing on shifts of the non-consensus base call frequencies,
events like loss of heterozygosity and clonal expansions in addition to somatic
variants can be detected. In the following, we will 1) outline the methodological
framework, 2) provide an example workflow on how to obtain a call set starting
from short-read alignments, and 3) illustrate potential benefits of the methods on
a biological cancer data set. This methodology can be used for a high
performance identification of variant sites as well as to quantitatively assess
the presence or absence in comparisons between matched samples.
2 Methodology
2.1 Comparative shift of non-consensus base call frequencies
Starting with a set of aligned reads we observe the sequencing depth \(N^{S}_{i}\) and the number of non-consensus base calls \(K^{S}_{i}\) for a sample \(S\) at position \(i\). Non-consensus is here defined as base calls that differ from the consensus sequence, which can be either the reference sequence or, in a comparative setting, the most abundant base call in the control sample. The non-consensus rate \(p^{S}_{i}\), which we assume to be binomially distributed, can then be estimated as
$$\hat{p}^{S}_{i} = \frac{K^{S}_{i}}{N^{S}_{i}}$$
for sites with \(N^{S}_{i} > 0\). Since \(K^{S}_{i} \leq N^{S}_{i}\), the rates are bound to the range \([0,1]\).
The true non-consensus rate
$$p^{S}_{i} = v^{S}_{i} + e^{S}_{i}$$
comprises the presence of a putative variant with a frequency \(v^{S}_{i}\) and a technical error rate \(e^{S}_{i}\). In order to detect and describe the change in the variant frequency, we focus on the shift \(d_{i}\) in non-consensus rates as the difference of the rates between the test and control samples, which we estimate as
$$\hat{d}_{i} = \hat{p}^{T}_{i} - \hat{p}^{C}_{i}.$$
If we assume that the true site-specific technical error rates are identical between the two matched samples [10], the difference of the rates yields an unbiased estimate for the change in the variant frequency. Thus, positions not haboring biological alterations will result in \(\hat{d}_{i} \approx 0\).
2.2 Confidence intervals
Distinguishing biological variants from noise requires knowledge about the variance of the point estimate \(\hat{d_{i}}\). By constructing a confidence interval (CI) for \(d_{i}\) with confidence level \(\beta\) [15], we assess the certainty of the estimated shift in non-consensus frequencies. The probability of the true value being outside the confidence interval is less than \(\alpha = 1 - \beta\). This is in concordance with the type I or \(\alpha\) error definition in statistical testing.
Under the assumption that the non-consensus counts \(K^{S}_{i}\) in our samples follow binomial distributions with parameters \(p^{S}_{i}\) and \(N^{S}_{i}\), several methods have been established for estimating confidence intervals for the difference of two rate parameters [13] [7]. The performance of an approach is generally described in term of their coverage probabilities indicating the probability of a confidence interval to cover the true value (see 5.3). Coverage probabilities greater and less than the confidence level \(\beta\) describe conservative and liberal behaviors, respectively. Due to the conservative coverage probabilities and high computational effort of exact confidence interval estimates, approximate methods are generally preferred [1] [7].
The Agresti-Caffo (AC) confidence interval [2]
$$\tilde{p}^{T} - \tilde{p}^{C} \pm z \sqrt{ \frac{\tilde{p}^{T} (1 - \tilde{p}^{T})} {\tilde{N}^{T}} + \frac{\tilde{p}^{C}(1 - \tilde{p}^{C})} {\tilde{N}^{C}} }$$
with
$$\tilde{p}^{X} = \frac{K^{X}+\zeta}{N^{X}+2\zeta},$$
$$\tilde{N}^{X} = N^{X} + 2\zeta,$$
$$\zeta = \frac{1}{4} z^2,$$
and \(z = z_{(1-\beta)/2}\) as the upper \((1-\beta)/2\) percentile of the standard normal distribution), is an approximation of the score test-based confidence interval. Several publications emphasize the usefulness and advantages of the AC method over related approaches [7] [3] [5].
2.2.1 Decision making with confidence intervals
While the estimate for the shift in the non-consenus frequency \(\hat{d}\) indicates the change in abundance and direction of a variant, the corresponding confidence interval gives us information about the precision and power of the estimate. Generally, wide confidence intervals will be present at sites with little statistical power, as due to low sequencing depths.
For the case that we compare a tumor to a matched normal sample, we shows a set of hypothetical cases that can be distinguished by regarding the point estimate and its confidence interval:
- Presence of a somatic, heterozygous variant
- Presence of a somatic, subclonal variant
- Presence of loss of heterozygosity
- Absence of a somatic variant
- Presence or absence of a variant cannot be distinguished due to the low certainty of the estimate
- No power due to insufficient sequencing depth

Illustrative cases of confidence intervals for somatic variant frequency estimates
2.3 Distinguishing event classes
Focusing on the comparative shift of non-consensus frequencies can detect and
distinguish different types of events. Since Rariant does not make explicit
assumptions about the abundance of a potential variant in the control sample, we
are further able to find clonal shifts, for example between different tumor
samples, or losses of heterozygocity. Generally, gains and losses of variant
alleles are characterized by positive and negative values of \(d\), respectively.
For a differentiated interpretation of the results, we classify a variants into
one of four classes:
- somatic
- A somatic variant that does not occur in the control sample
- hetero/LOH
- A shift away from heterozygous SNP in the control sample
- undecided
- Both of the 'somatic' or 'hetero' are possible
- powerless
- A distinction between the two classes cannot be made due to a lack of power
The classification is based on two binomial tests for each position:
- Somatic variants where the variant allele is not present in the control sample, rejecting a binomial test with the alternative hypothesis \(H_{1}: p^{C} > 0\).
- Sites with a loss of heterozygosity with a shift away from a heterozygous variant in the control sample, rejecting a binomial test with the alternative hypothesis \(H_{1}: p^{C} \neq \frac{1}{2}\).
2.4 Identifying variant sites in large datasets
The method that we have described before is suited for detecting variant positions efficiently in large sequencing datasets, including whole-exome and whole-genome sequencing. For this purpose, we test for a shift in non-consensus frequencies between two samples at each genomic position individually:
- Form the base counts table for four bases A, C, G, T from the aligned reads. In order to reduce the number of false counts, we can optionally exclude reads with low base calling quality and clip the head of each read.
- Determine the consensus sequence: In a comparative setting, we will use the most abundant base call.
- Calculate the sequencing depth \(N^{S}_{i}\), mismatch counts \(K^{S}_{i}\), and derived statistics for both samples, based on the consensus sequence (see 2.1).
- Find potential variant sites with a Fisher's Exact Test, comparing the number of mismatching and total bases between the samples: \({K^{T}_{i}, N^{T}_{i}, K^{C}_{i}, N^{C}_{i}}\). The p-values are corrected for multiple testing according to the Benjamini-Hochberg procedure. Only positions rejecting the null hypothesis at a significance level \(\alpha\) are furtheron considered as potential variants.
- Calculate Agresti-Caffo confidence intervals with confidence level \(\beta\), in order to evaluate presence or absence of the variant (see 2.2).
- Classify variant sites into the groups: somatic, LOH, undecided, and powerless (see 2.3).
3 Workflow
In the following, we show a complete workflow for identifying SNVs from aligned
short reads. For illustration purposes, we will focus on data from a whole
exome sequencing (WES) study, as part of the h5vcData package
[12], covering a subset of the NRAS gene.
library(Rariant) library(h5vcData) library(GenomicRanges) library(ggbio) library(ggplot2)
3.1 The data set
We compare an AML tumor sample with the matching control sample of a single
patient, starting with the alignments stored in BAM files. Here, we will use
the system.file function to construct the path to our example data files.
control_bam = system.file("extdata", "NRAS.Control.bam", package = "h5vcData", mustWork = TRUE) test_bam = system.file("extdata", "NRAS.AML.bam", package = "h5vcData", mustWork = TRUE)
Since we restrict our analysis to a small region of the genome at the moment, we further define our region of interest.
roi = GRanges("1", IRanges(start = 115258439, end = 115259089))
3.2 Identifying variant sites
Variant sites can be identified with the rariant function. As input, we
specify the alignment files for the test and control sample. In case that we
are only interested in calling variants in specific regions, we can pass a
GRanges object with the given intervals as the region argument. Otherwise,
if this argument is omitted, the entire genome will be analyzed.
vars = rariant(test_bam, control_bam, roi)
The calls are returned as a GRanges object, with each row corresponding to a
detected variant site. In this case, one variant is classified as a probable
somatic variant, with an estimated shift d in the variant frequency of \(\approx
0.51\) within the \(95%\) confidence interval \([0.37,0.62]\).
vars
GRanges with 1 range and 24 metadata columns:
seqnames ranges strand | testMismatch controlMismatch testDepth controlDepth testRef testAlt
|
[1] 1 [115258747, 115258747] * | 32 0 63 81 G G
controlRef ref refDepth altDepth called p1 p2 d ds lower upper
[1] C C 31 32 TRUE 0.5079 0 0.5079 0.4961 0.3723 0.6199
pval padj eventType padjSomatic padjHetero pvalSomatic pvalHetero
[1] 8.577e-15 5.447e-12 somatic 1 8.272e-25 1 8.272e-25
---
seqlengths:
1
249250621
Additional arguments allow us to change the confidence levels and the filter settings used for excluding low quality base calls to reduce false positives. The defaults are suited for current Illumina sequencing data sets.
3.3 Interpreting the results
The columns of the GRanges object returned by rariant summarizes the
evidence for the presence or absence of a variant:
- testMismatch, controlMismatch
- Non-consensus base counts \(K_{i}^{T}\) and \(K_{i}^{C}\) in the test and control sample
- testDepth, controlDepth
- Sequencing depth \(N_{i}^{T}\) and \(N_{i}^{C}\) in the test and control sample
- testRef, controlRef
- Most abundant base call in the test and control sample,
with
Nrefering to multiple ones. - testAlt
- Most abundant mismatch/non-consensus base call, with
Nrefering to multiple ones. - ref
- Consensus sequence
- d, ds
- Estimated shift \(\hat{d}_{i}\) of the non-consensus frequencies, with
dsas the shrinkage estimate - p1, p2
- Non-consensus rates \(p_{i}^{T}\) and \(p_{i}^{C}\) in the test and control sample
- lower, upper
- Lower and upper bound of the confidence interval for \(d\)
- pval, padj
- Raw and Benjamini-Hochberg adjusted p-value of the Fisher's Exact test
- called
- Was the site called as a variant?
- eventType
- Type of variant event: 'somatic', 'loh', 'undecided'.
- pvalSomatic, padjSomatic, pvalHetero, padjHetero
- Raw and Benjamini-Hochberg adjusted p-values of the binomial tests for the respective event types
3.4 Exploratory variant analysis
By default, only identified variants are returned. We can also obtain the
results for all sites in our region of interest with select = FALSE. This
will be useful for an exploratory analysis, such as investigating the absence of
a variant or comparing calls between samples.
vars_all = rariant(test_bam, control_bam, roi, select = FALSE) head(vars_all, 3)
GRanges with 3 ranges and 24 metadata columns:
seqnames ranges strand | testMismatch controlMismatch testDepth controlDepth testRef testAlt
|
[1] 1 [115258439, 115258439] * | 0 0 1 1 A N
[2] 1 [115258440, 115258440] * | 0 0 1 1 G N
[3] 1 [115258441, 115258441] * | 0 0 0 1 N N
controlRef ref refDepth altDepth called p1 p2 d ds lower upper
[1] A A 1 FALSE 0 0 0 0.0000 -0.7619 0.7619
[2] G G 1 FALSE 0 0 0 0.0000 -0.7619 0.7619
[3] T T 0 FALSE 0 0.1712
pval padj eventType padjSomatic padjHetero pvalSomatic pvalHetero
[1] 1 1 powerless 1 1 1 1
[2] 1 1 powerless 1 1 1 1
[3] 1 1 powerless 1 1 1 1
---
seqlengths:
1
249250621
3.5 Summarizing and visualizing results
Sites harboring potential biological variants can be identified by confidence
intervals that reject non-consensus frequencies shifts of 0. The ciOutside
function finds sites whose confidence intervals do not overlap a value of
interest. As we have seen before, the NRAS locus contains one such site.
idx_out = ciOutside(vars_all, 0) ind_out = which(idx_out) vars_all$outside = idx_out table(idx_out)
idx_out
FALSE TRUE
634 1
We inspect the variant site by visualizing the confidence intervals. This allows us to clearly identify the variant and quantify the range of the expected variant frequency, as well as state the absence of other variants in the surrounding with high certainty. The second plot indicates the shift in relation to the estimates \(p^{T}_{i}\) and \(p^{C}_{i}\), also indicating the gain of the variant allele in the tumor.
win = 20 ind_var = (ind_out[1] - win):(ind_out[1] + win) p_ci = plotConfidenceIntervals(vars_all[ind_var]) p_shift = plotAbundanceShift(vars_all[ind_var]) t = tracks(p_ci, p_shift) print(t)

NRAS: Variant frequency confidence intervals and shifts. Red and blue points show the non-consensus rates in the test and control sample. The difference between the two represents the effect size.
Looking at a larger region, we see that the certainty of our estimates correlates with sequencing depth of the samples. We describe this relationship in more detail in the supplementary section 5.2.
ind_low = (100 - 40):(100 + 40) p_low = plotConfidenceIntervals(vars_all[ind_low]) p_depth = autoplot(vars_all[ind_low], aes(y = testDepth), geom = "step", col = "darkred") + geom_step(aes(y = controlDepth), col = "steelblue3") + theme_bw() t2 = tracks(p_low, p_depth) print(t2)

NRAS: Non-variant site with sequencing depth
3.6 Interactive variant analysis
With the rariantInspect interface, the results of the rariant can be
explored interactively in the web browser. Since we cannot demonstrate this in
a static document, we show screenshots of the application. Figures and results
tables can be displayed conveniently and subset according to multiple criteria.
rariantInspect(vars_all)
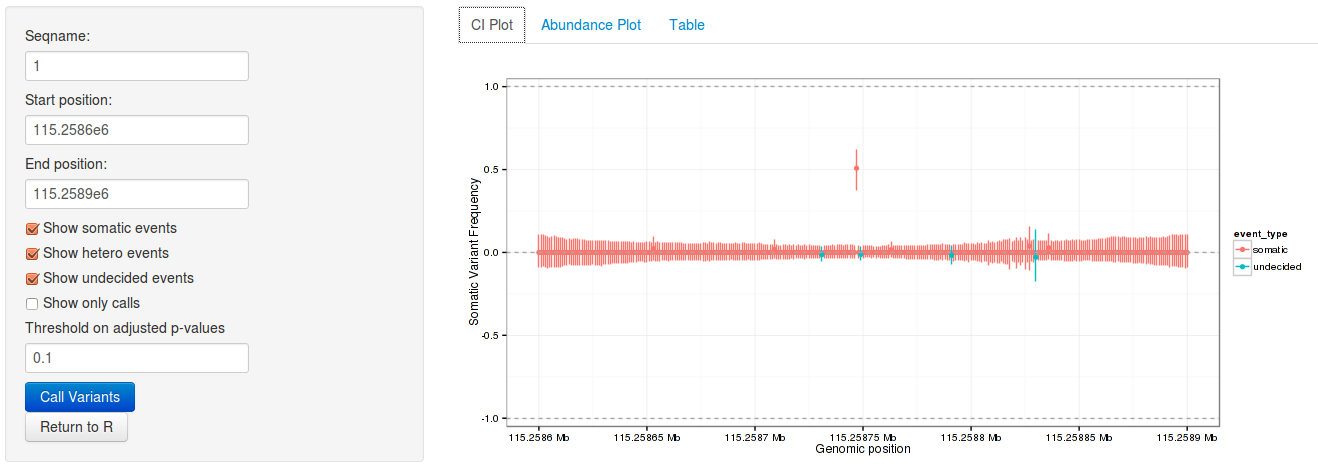
Figure 1: Interactive analysis, showing confidence interval plots
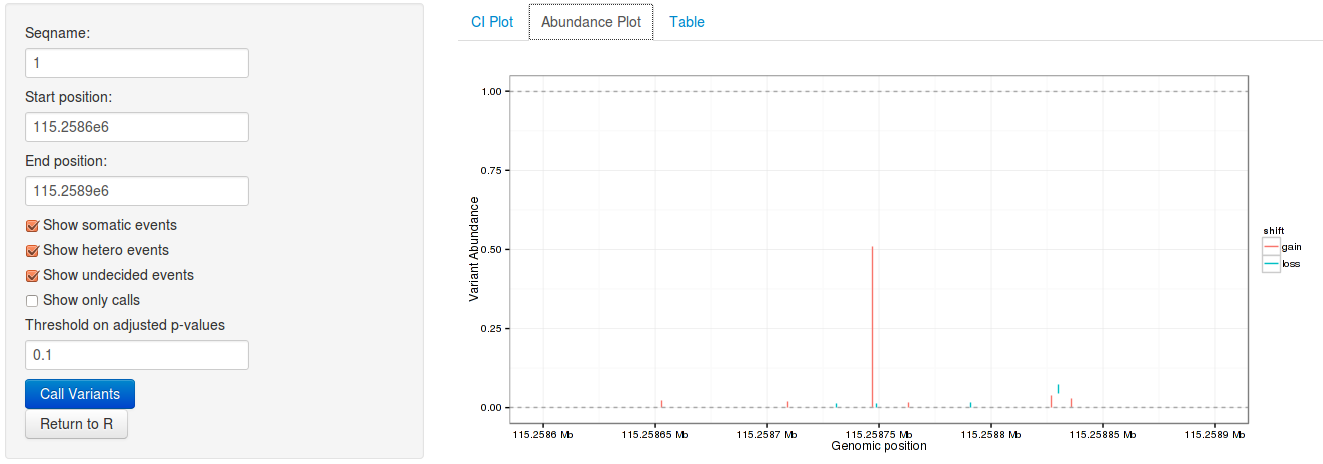
Figure 2: Interactive analysis, showing non-consensus rate shift plots
4 Example Cases
4.1 1000 Genomes Simulation Study
We want to further demostrate the usage and abilities of the Rariant package
on a real-life data set. Due to legal and privacy issues, most human cancer
sequencing data is not publicly accessible and therefore cannot serve as an
example data set here. Alternatively, we conduct an analysis to mimic the
characteristics of current cancer sequencing studies.
For the purpose of the analysis, we compare three samples from the 1000 Genomes
project [6], serving as a
control/normal (control) and two related test/tumor samples (test and
test2). Further, we simulate a clonal mixture (mix) of the two test samples
by combining their reads.
library(Rariant) library(GenomicRanges) library(ggbio)
tp53_region = GRanges("chr17", IRanges(7571720, 7590863))
control_bam = system.file("extdata", "control.bam", package = "Rariant", mustWork = TRUE) test1_bam = system.file("extdata", "test.bam", package = "Rariant", mustWork = TRUE) test2_bam = system.file("extdata", "test2.bam", package = "Rariant", mustWork = TRUE) mix_bam = system.file("extdata", "mix.bam", package = "Rariant", mustWork = TRUE)
v_test1 = rariant(test1_bam, control_bam, tp53_region, select = FALSE) v_test2 = rariant(test2_bam, control_bam, tp53_region, select = FALSE) v_mix = rariant(mix_bam, control_bam, tp53_region, select = FALSE)
In the following, we look at positions which showed a significant effect in at least one sample. This gives us 12 positions to consider in the following.
poi = unique(c(v_test1[ciOutside(v_test1)], v_test2[ciOutside(v_test2)], v_mix[ciOutside(v_mix)])) length(poi)
[1] 12
head(poi)
GRanges with 6 ranges and 24 metadata columns:
seqnames ranges strand | testMismatch controlMismatch testDepth controlDepth testRef testAlt
|
[1] chr17 [7574775, 7574775] * | 18 0 37 41 C T
[2] chr17 [7574779, 7574779] * | 19 0 40 44 C T
[3] chr17 [7574936, 7574936] * | 20 0 36 30 T T
[4] chr17 [7575564, 7575564] * | 17 0 31 34 T T
[5] chr17 [7578115, 7578115] * | 18 0 33 42 T T
[6] chr17 [7578645, 7578645] * | 18 0 33 20 C C
controlRef ref refDepth altDepth called p1 p2 d ds lower upper
[1] C C 19 18 TRUE 0.4865 0 0.4865 0.4648 0.3016 0.6279
[2] C C 21 19 TRUE 0.4750 0 0.4750 0.4552 0.2985 0.6120
[3] C C 16 20 TRUE 0.5556 0 0.5556 0.5227 0.3537 0.6916
[4] C C 14 17 TRUE 0.5484 0 0.5484 0.5188 0.3407 0.6969
[5] C C 15 18 TRUE 0.5455 0 0.5455 0.5211 0.3503 0.6919
[6] T T 15 18 TRUE 0.5455 0 0.5455 0.4991 0.3130 0.6853
pval padj eventType padjSomatic padjHetero pvalSomatic pvalHetero
[1] 8.314e-08 3.183e-04 somatic 1 2.268e-12 1 9.095e-13
[2] 3.974e-08 1.902e-04 somatic 1 4.308e-13 1 1.137e-13
[3] 2.288e-07 5.476e-04 somatic 1 2.123e-09 1 1.863e-09
[4] 1.420e-07 4.182e-04 somatic 1 1.549e-10 1 1.164e-10
[5] 1.085e-08 6.922e-05 somatic 1 1.290e-12 1 4.547e-13
[6] 1.740e-05 3.331e-02 somatic 1 1.929e-06 1 1.907e-06
---
seqlengths:
chr17
81195210
v_poi1 = v_test1[v_test1 %in% poi] v_poi2 = v_test2[v_test2 %in% poi] v_poi3 = v_mix[v_mix %in% poi]
To get a better understanding about the evidence for the presence or absence of particular variants acros samples, we plot the confidence intervals, colored according to the predicted event type, and abundance shifts for all sites of interest, colored according to the sign of the shift.
py1 = plotConfidenceIntervals(v_poi1, color = "eventType") py2 = plotConfidenceIntervals(v_poi2, color = "eventType") py3 = plotConfidenceIntervals(v_poi3, color = "eventType") t = tracks(py1, py2, py3) print(t)

Confidence intervals for simulition study
While most of the variants are somatic, i.e. they do not appear in the control sample, the last variant position shows a loss of a heterozygous SNP. Looking for example in more detail into the group of 5 variant sites around 7.85 Mbp: We can identify them as consistent with a heterozygous somatic variant in the first sample, since their 95% CIs overlap the value of 0.5. In contrast, we can show the absence of the same variants in the second sample. The third sample again shows the presence of the variants, as seen in the first case, but with lower abundance. Such a result could be expected in a mixture of subclones, in which some clones carry a somatic variant and others not. Further, we can also see the case of the next variant which consistently exists in all three samples with the same abundance.
pa1 = plotAbundanceShift(v_poi1) pa2 = plotAbundanceShift(v_poi2) pa3 = plotAbundanceShift(v_poi3) t2 = tracks(pa1, pa2, pa3) print(t2)

Abundance shifts for simulition study
4.2 AML Genome Sequencing
To illustrate typical cases that can be distinguished with the proposed methodology
on real data, we investigate a tumor/normal comparison of a single patient as
part of an AML WGS study. The data is part of the h5vcData package
[12]. We will focus on the APOBEC3A locus on chromosome
22, and will use two types of plots of a set of exemplary regions:
- Mismatch plots which show the sequencing depth (in gray) and base-specific
mismatches (in colors) separated across strands, with the normal in the top
and the tumor sample in the bottom panel. Positive and negative values
correspond to the plus and minus strand, respectively. The plots are
generated with the
h5vcpackage [18], and details on how to generate these are explained in the package vignette. - Confidence interval plot with the estimated somatic variant frequency (as
dot) and corresponding 99% confidence interval (as line range) for both as
well as the plus and minus strand. The plots are generated with the
plotConfidenceIntervalsfunction.
4.2.1 Case 1: Somatic variant
The first example shows a potential heterozygous somatic variant, for which there is no evidence in the control sample. While the mismatch frequencies differ slightly between the two strands, the overlap of the two confidence intervals indicate that there is no disagreement.

Mismatch plot for case 1: Somatic variant

Confidence interval plot for case 1: Somatic variant
4.2.2 Case 2: Absence of variants
In contrast to the previous case does this region not show a somatic variant. The confidence intervals are all consistent with a somatic variant frequency of 0, while the small width of them indicate the high certainty of the estimate and of the call of absence.

Mismatch plot for case 2: Absence of a variant

Confidence interval plot for case 2: Absence of a variant
4.2.3 Case 3: Strand-specific mismatches
In the third example region, mismatches are predominantly present on the minus strand. This behaviour is also reflected in the confidence intervals, comparing the plus and the minus strand to each other.

Mismatch plot for case 3: Strand-specific mismatches

Confidence interval plot for case 3: Strand-specific mismatches
4.2.4 Case 4: Strand-specific differences in sequencing depth
The fourth region denotes a case with deviating sequencing depth between the strands, which can be observed at the border of exons in exon-sequencing datasets. For the plus strand, the low statistical power is reflected in the wide confidence intervals.

Mismatch plot for case 4: Strand-specific differences in sequencing depth

Confidence inteval plot for case 4: Strand-specific differences in sequencing depth
5 Supplementary Information
5.1 Strand-specific analysis
By comparing the confidence intervals between strands, we can further detect and characterize effects such as variations in sequencing depth and strand biases. We illustrate this with a set of hypothetical cases for confidence intervals for two strands. The upper row (cases 4-7) corresponds to sites with overlapping CIs, whereas the lower row (cases 1-3) shows cases of disagreements between the CIs indicative of strand biases. When analyzing the probability for the overlap of confidence intervals, an adjustment of the confidence level has to be taken into account [8].

Illustrative cases of confidence intervals for somatic variant frequency estimates for two strands
Motivated by the analysis of different Illumina genome and exome sequencing, we consider strand-biases, in which the non-consensus base call rates differ significantly between strands at sites with sufficient sequencinq depth, a neglectable problem with current data sets and analysis pipelines (see also Best practices for short-read processing). In the presence of strand biases, pooling the counts of both plus and minus strand may be not desirable. A possible solution may be to perform a strand-specific analysis, and later combine the resulting statistics. Gerstung and colleagues discuss different approaches for combining p-values [9], in particular taking the minimum, maximum, average, or Fisher combination. These can be also applied for confidence intervals, with Fisher's method being equivalent to taking the sum of both strands.
5.2 Statistical power and sequencing depth
The statistical power, and thereby the width of the confidence interval, depends on the sequencing depths in both samples. For the region harboring the variant site, we can illustrate the relationship between by plotting the confidence interval width against the sequencing depth averaged over both samples.
df = as.data.frame(vars_all) df$ci_width = ciWidth(df) p = ggplot(df) + geom_point(aes_string(x = "(controlDepth + testDepth) / 2", y = "ci_width", col = "outside")) + xlab("Average sequencing depth") + ylab("Confidence interval width") + theme_bw() print(p)

Confidence interval width - sequencing depth relationship. The identified variant is marked in blue.
5.3 Assessing performance of confidence interval methods
As outlined before, an important property for assessing confidence intervals is given by their coverage probabilities. Ideally, we would expect a method to have coverage probabilities close to the nominal confidence level β over a wide range in the parameter space. Previous publications analyzing the performance focus on parameter settings that deviate from those of sequencing data sets [7]. Therefore, we perform a simulation that demonstrates the behavior of the Agrest-Caffo methods for a whole-genome sequencing study. For a fixed sequencing depth of 30 in both test and control sample, the coverage probability of 95% AC confidence intervals is computed for all possible combinations of mismatch counts \(K^{T}\) and \(K^{C}\).
## WGS n1 = 30 n2 = 30 k1 = 0:(n1 - 1) k2 = 0:(n2 - 1) cl = 0.95 n_sample = 10000 pars = expand.grid(k1 = k1, k2 = k2, n1 = n1, n2 = n2, conf_level = cl) cp_ac = coverageProbability(pars, fun = acCi, n_sample = n_sample)
p_ac = ggplot(cp_ac) + geom_tile(aes(x = k1, y = k2, fill = cp)) + scale_fill_gradient2(midpoint = 0.95, limits = c(0.9, 1)) + theme_bw() + xlab("kT") + ylab("kC") print(p_ac)

Coverage probabilities for whole-genome setting
For mismatch rates close to 0 or 1 in both samples, the Agresti-Caffo method shows a conservative perfomance.
5.4 Benchmarking of performance and resources
For an analysis of two matched human tumor samples, we performed a benchmark to
assess the computational time and memory usage on a standard laptop (Thinkpad
X220 built in 2011). Both samples contain about 95M reads mapped to the
1000genomes reference sequence reads that are considered in the analysis. For
the analysis of chromosome 22, the analysis with default parameters required
~873s and 600MB of RAM. For an analysis of all linear toplevel chromosomes
(autosomes and allosomes), this would require ~15h of time. Please consider
that the current version of Rariant is under active development and
computational efficiency will increase with newer versions.
6 Frequently Asked Questions
6.1 Getting help
We welcome emails with questions or suggestions about our software, and want to ensure that we eliminate issues if and when they appear. We have a few requests to optimize the process:
- All emails and follow-up questions should take place over the Bioconductor mailing list, which serves as a repository of information.
- The subject line should contain Rariant and a few words describing the problem. First search the Bioconductor mailing list, for past threads which might have answered your question.
- If you have a question about the behavior of a function, read the sections of
the manual page for this function by typing a question mark and the function
name, e.g.
?rariant. Additionally, read through the vignette to understand the interplay between different functions of the package. We spend a lot of time documenting individual functions and the exact steps that the software is performing. - Include all of your R code related to the question you are asking.
- Include complete warning or error messages, and conclude your message with the
full output of
sessionInfo().
6.2 Installing the package
Before you want to install the Rariant package, please ensure that
you have the latest version of R and Bioconductor installed. For details on
this, please have a look at the help packages for R and Bioconductor. Then you
can open R and run the following commands which will install the latest
release version of Rariant:
source("http://bioconductor.org/biocLite.R") biocLite("Rariant")
7 References
| [1] | Alan Agresti and Brent A. Coull. Approximate is better than exact for interval estimation of binomial proportions. The American Statistician, 52(2):119126, 1998. [ http ] |
| [2] | Alan Agresti and Brian Caffo. Simple and effective confidence intervals for proportions and differences of proportions result from adding two successes and two failures. The American Statistician, 54(4):280288, 2000. [ http ] |
| [3] | Walter W. Piegorsch. Sample sizes for improved binomial confidence intervals. Computational Statistics & Data Analysis, 46(2):309-316, June 2004. [ DOI | http ] |
| [4] | Yoav Benjamini and Daniel Yekutieli. False discovery rateadjusted multiple confidence intervals for selected parameters. Journal of the American Statistical Association, 100(469):7181, 2005. [ http ] |
| [5] | Frank Schaarschmidt, Martin Sill, and Ludwig A. Hothorn. Approximate simultaneous confidence intervals for multiple contrasts of binomial proportions. Biometrical Journal, 50(5):782792, 2008. [ DOI | http ] |
| [6] | The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature, 467(7319):1061-1073, October 2010. [ DOI | .html ] |
| [7] | Morten W. Fagerland, Stian Lydersen, and Petter Laake. Recommended confidence intervals for two independent binomial proportions. Statistical methods in medical research, 2011. [ http ] |
| [8] | Mirjam J. Knol, Wiebe R. Pestman, and Diederick E. Grobbee. The (mis)use of overlap of confidence intervals to assess effect modification. European Journal of Epidemiology, 26(4):253-254, April 2011. PMID: 21424218 PMCID: PMC3088813. [ DOI | http ] |
| [9] | Moritz Gerstung, Christian Beisel, Markus Rechsteiner, Peter Wild, Peter Schraml, Holger Moch, and Niko Beerenwinkel. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nature Communications, 3:811, May 2012. [ DOI | .html ] |
| [10] | Omkar Muralidharan, Georges Natsoulis, John Bell, Hanlee Ji, and Nancy R. Zhang. Detecting mutations in mixed sample sequencing data using empirical bayes. The Annals of Applied Statistics, 6(3):1047-1067, September 2012. Zentralblatt MATH identifier06096521, Mathematical Reviews number (MathSciNet) MR3012520. [ DOI | http ] |
| [11] | Lucy R. Yates and Peter J. Campbell. Evolution of the cancer genome. Nature Reviews Genetics, 13(11):795, November 2012. [ DOI | .html ] |
| [12] | Paul Theodor Pyl. h5vcData: example data for the h5vc package, 2013. [ .html ] |
| [13] | Joseph L. Fleiss, Bruce Levin, and Myunghee Cho Paik. Statistical methods for rates and proportions. John Wiley & Sons, 2013. [ http ] |
| [14] | Geoffrey Decrouez and Peter Hall. Split sample methods for constructing confidence intervals for binomial and poisson parameters. Journal of the Royal Statistical Society: Series B (Statistical Methodology), page n/an/a, 2013. [ DOI | http ] |
| [15] | Alan Agresti. Categorical data analysis. Wiley, Hoboken, NJ, 2013. |
| [16] | Su Y. Kim and Terence P. Speed. Comparing somatic mutation-callers: beyond venn diagrams. BMC Bioinformatics, 14(1):189, June 2013. PMID: 23758877. [ DOI | http ] |
| [17] | Nicola D. Roberts, R. Daniel Kortschak, Wendy T. Parker, Andreas W. Schreiber, Susan Branford, Hamish S. Scott, Garique Glonek, and David L. Adelson. A comparative analysis of algorithms for somatic SNV detection in cancer. Bioinformatics, 29(18):2223-2230, September 2013. PMID: 23842810. [ DOI | http ] |
| [18] | Paul Theodor Pyl, Julian Gehring, Bernd Fischer, and Wolfgang Huber. h5vc: Scalable nucleotide tallies with HDF5. Bioinformatics, page btu026, January 2014. PMID: 24451629. [ DOI | http ] |
8 Session Info
R version 3.1.0 (2014-04-10)
Platform: x86_64-unknown-linux-gnu (64-bit)
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8 LC_COLLATE=C
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8 LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid parallel stats graphics grDevices utils datasets methods base
other attached packages:
[1] biovizBase_1.12.0 h5vc_1.2.0 gridExtra_0.9.1 XVector_0.4.0 ggbio_1.12.0
[6] GenomicRanges_1.16.0 GenomeInfoDb_1.0.0 IRanges_1.21.45 h5vcData_1.0.1 Rariant_1.0.0
[11] bigmemory_4.4.6 BH_1.54.0-1 bigmemory.sri_0.1.2 Biobase_2.24.0 BiocGenerics_0.10.0
[16] ggplot2_0.9.3.1 knitr_1.5
loaded via a namespace (and not attached):
[1] AnnotationDbi_1.26.0 BBmisc_1.5 BSgenome_1.32.0 BatchJobs_1.2
[5] BiocParallel_0.6.0 Biostrings_2.32.0 DBI_0.2-7 Formula_1.1-1
[9] GenomicAlignments_1.0.0 GenomicFeatures_1.16.0 Hmisc_3.14-3 MASS_7.3-31
[13] NMF_0.20.5 RColorBrewer_1.0-5 RCurl_1.95-4.1 RJSONIO_1.0-3
[17] RSQLite_0.11.4 Rcpp_0.11.1 Rsamtools_1.16.0 SomaticSignatures_1.0.0
[21] VariantAnnotation_1.10.0 XML_3.98-1.1 abind_1.4-0 biomaRt_2.20.0
[25] bitops_1.0-6 brew_1.0-6 caTools_1.16 cluster_1.15.2
[29] codetools_0.2-8 colorspace_1.2-4 dichromat_2.0-0 digest_0.6.4
[33] doParallel_1.0.8 evaluate_0.5.3 exomeCopy_1.10.0 fail_1.2
[37] foreach_1.4.2 formatR_0.10 gridBase_0.4-7 gtable_0.1.2
[41] gtools_3.3.1 highr_0.3 httpuv_1.3.0 iterators_1.0.7
[45] labeling_0.2 lattice_0.20-29 latticeExtra_0.6-26 markdown_0.6.5
[49] munsell_0.4.2 pcaMethods_1.54.0 pkgmaker_0.20 plyr_1.8.1
[53] proto_0.3-10 registry_0.2 reshape_0.8.4 reshape2_1.2.2
[57] rhdf5_2.8.0 rngtools_1.2.4 rtracklayer_1.24.0 scales_0.2.3
[61] sendmailR_1.1-2 shiny_0.9.1 splines_3.1.0 stats4_3.1.0
[65] stringr_0.6.2 survival_2.37-7 tools_3.1.0 xtable_1.7-3
[69] zlibbioc_1.10.0